Genome Res. 14:1832-1850, 2004
©2004 by
Cold Spring Harbor Laboratory Press; ISSN 1088-9051/04 $5.00
Letter
Mitochondrial Genome Variation in Eastern Asia and the Peopling of
Japan Masashi Tanaka1,15,
Vicente M. Cabrera2, Ana M.
González2, José M. Larruga2,
Takeshi Takeyasu1,3, Noriyuki
Fuku1,4, Li-Jun
Guo1,3, Raita
Hirose1, Yasunori Fujita1,
Miyuki Kurata1, Ken-ichi
Shinoda5, Kazuo Umetsu6,
Yoshiji Yamada7,1, Yoshiharu
Oshida3, Yuzo Sato3,
Nobutaka Hattori8, Yoshikuni
Mizuno8, Yasumichi Arai10,
Nobuyoshi Hirose10, Shigeo
Ohta11, Osamu Ogawa9,
Yasushi Tanaka9, Ryuzo
Kawamori9, Masayo
Shamoto-Nagai1,4,12, Wakako
Maruyama12, Hiroshi Shimokata13,
Ryota Suzuki14 and Hidetoshi
Shimodaira14
1 Department of Gene Therapy, Gifu International
Institute of Biotechnology, Kakamigahara, Gifu 504-0838, Japan ,
2 Department of Genetics, Faculty of Biology, University of
La Laguna, Tenerife 38271, Spain , 3 Department of
Sports Medicine, Graduate School of Medicine, Nagoya University, Nagoya
464-8601, Japan , 4 Japan Science and Technology Agency,
Kawaguchi, Saitama 332-0012, Japan , 5 Department of
Anthropology, National Science Museum, Tokyo 169-0073, Japan ,
6 Department of Forensic Medicine, Yamagata University
School of Medicine, Yamagata 990-9585, Japan , 7
Department of Human Functional Genomics, Life Science Research Center,
Mie University, Tu-shi, Mie 514-8507, Japan , 8
Department of Neurology, Metabolism and Endocrinology, Juntendo
University School of Medicine, Tokyo 113-8421, Japan , 9
Department of Medicine, Metabolism and Endocrinology, Juntendo
University School of Medicine, Tokyo 113-8421, Japan , 10
Department of Geriatric Medicine, Keio University School of Medicine,
Tokyo 160-8582, Japan , 11 Department of Biochemistry
and Cell Biology, Institute of Gerontology, Nihon Medical School, Kawasaki
211-8533, Japan , 12 Laboratory of Biochemistry and
Metabolism, Department of Basic Gerontology, National Institute for
Longevity Sciences, Obu 474-8522, Japan , 13 Department
of Epidemiology, National Institute for Longevity Sciences, Obu 474-8522,
Japan , 14 Department of Mathematical and Computing
Sciences, Tokyo Institute of Technology, Tokyo 152-8552, Japan
![]() |
ABSTRACT |
To construct
an East Asia mitochondrial DNA (mtDNA) phylogeny, we sequenced
the complete mitochondrial genomes of 672 Japanese individuals
(http://www.giib.or.jp/mtsnp/index_e.html).
This allowed us to perform a phylogenetic analysis with a pool
of 942 Asiatic sequences. New clades and subclades emerged
from the Japanese data. On the basis of this unequivocal
phylogeny, we classified 4713 Asian partial mitochondrial
sequences, with <10% ambiguity. Applying population and
phylogeographic methods, we used these sequences to shed light
on the controversial issue of the peopling of Japan.
Population-based comparisons confirmed that present-day
Japanese have their closest genetic affinity to northern Asian
populations, especially to Koreans, which finding is congruent
with the proposed Continental gene flow to Japan after the
Yayoi period. This phylogeographic approach unraveled a high
degree of differentiation in Paleolithic Japanese. Ancient
southern and northern migrations were detected based on the
existence of basic M and N lineages in Ryukyuans and Ainu.
Direct connections with Tibet, parallel to those found for the
Y-chromosome, were also apparent. Furthermore, the highest
diversity found in Japan for some derived clades suggests that
Japan could be included in an area of migratory expansion to
Continental Asia. All the theories that have been proposed up
to now to explain the peopling of Japan seem insufficient to
accommodate fully this complex picture.
Recent analysis of global mitochondrial DNA diversity in humans
based on complete mtDNA sequences has provided compelling
evidence of a human mtDNA origin in Africa (Ingman et al.
2000![Go]() ). Less than
100,000 years ago, at least two mtDNA human lineages began to
rapidly spread from Africa to the Old World (Maca-Meyer et al.
2001 ). Less than
100,000 years ago, at least two mtDNA human lineages began to
rapidly spread from Africa to the Old World (Maca-Meyer et al.
2001 ). The archaeological records attest that humans reached
Japan, at the eastern edge of Asia, around 30,000 years ago
(Glover 1980). At that time, Japan was connected to the Continent
by both northern and southern land bridges, enabling two
migratory routes. As early as 13,000 years ago, pottery
appeared in Japan and Siberia for the first time in the world
(Shiraishi 2002). Subsequent technical improvements gave rise to
the Japanese Neolithic period known as the Jomon period, in
which the population growth was considerable. Later,
Continental people arrived in Japan from the Korean peninsula,
initiating the Yayoi period, with this migration reaching its
maximum at the beginning of the first millennium. ). The archaeological records attest that humans reached
Japan, at the eastern edge of Asia, around 30,000 years ago
(Glover 1980). At that time, Japan was connected to the Continent
by both northern and southern land bridges, enabling two
migratory routes. As early as 13,000 years ago, pottery
appeared in Japan and Siberia for the first time in the world
(Shiraishi 2002). Subsequent technical improvements gave rise to
the Japanese Neolithic period known as the Jomon period, in
which the population growth was considerable. Later,
Continental people arrived in Japan from the Korean peninsula,
initiating the Yayoi period, with this migration reaching its
maximum at the beginning of the first millennium.
With this archaeological framework in mind, it was of
anthropological interest to us to know whether the modern
Japanese are the result of an admixture between the
Paleolithic-Neolithic aborigines and more recent immigrant
populations, whether the indigenous population gradually
evolved to give rise to the modern Japanese, with subsequent
colonizations having strong cultural influences but only minor
demographic impact, or even whether the late Neolithic waves
entirely replaced the indigenous residents. Morphometric data
obtained from the remains of Japanese Paleolithic people are
more in accordance with a southern origin for these first
immigrants. Subsequent morphological studies on modern
indigenous (northern Ainu and southern Ryukyuans) and mainland
Japanese favored an admixture model in which the former would
be descendants of the Paleolithic Japanese and the latter
derived from the Continental immigrants who gave rise to the
Yayoi period (Hanihara 1991). Genetic analysis using classical markers assigned
a definitive northern origin to the Upper Paleolithic
inhabitants of Japan; but whereas some authors favored a
homogeneous background for all modern Japanese (Nei 1995), others claimed that although Upper Paleolithic
and Yayoi period immigrants had probably a northern Asian
origin, they were genetically differentiated (Omoto and Saitou
1997). The application of molecular markers to define
maternal and paternal lineages to the peopling of Japan
confirmed the dual admixture model but added some interesting
novelties. For example, the study of Y-chromosome markers led
to the discovery of remarkable Korean and Tibetan influences
on the Japanese population (Hammer and Horai 1995); and mtDNA HVS-I sequences also confirmed the
Korean input (Horai et al. 1996) and closer affinities of the Japanese to Tibetans than
to southern Asians (Qian et al. 2001). In quantitative estimations of maternal
admixture, it was found that  65% of the mainland
Japanese gene pool was derived from Continental gene flow after
the Yayoi period. However, the indigenous Ainu from the
northern island of Hokkaido and the Ryukyuans from southern
Okinawa showed <20% Continental specificity, pointing to
them as the most probable descendants of the Jomon people. The
fact that these indigenous groups were, in turn, genetically
well differentiated indicated a notable degree of heterogeneity
and/or isolation among the early Japanese immigrants (Horai et
al. 1996). However, two handicaps of these studies are the
incomplete representation of Asian populations and the
relatively small sample size of those analyzed, which weakens
the reliance on the relative affinities found by genetic
distance methods (Helgason et al. 2001). For mtDNA there are currently enough HVI/HVII
data from eastern Asia, including Japan, to test the validity
of the above-mentioned results. However, these sequences have
been assorted into different clades following different
insufficient criteria or even have not been classified at all.
Furthermore, the phylogenetic confidence of results based only
on sequences from the noncoding region (HVI, HVII) has been
recently questioned (Bandelt et al. 2000). This is mainly due to the frequent occurrence
of parallel mutations in independent lineages that confuse the
correct classification, a source of error that is increased
because the basal motif in the noncoding region for the two
macrolineages that expanded throughout Asia is the same
(16223). In addition, as the noncoding region has not evolved
at a constant rate across all human lineages, it is considered
inappropriate to use this region for dating evolutionary events
(Ingman et al. 2000; Finnilä et al. 2001). 65% of the mainland
Japanese gene pool was derived from Continental gene flow after
the Yayoi period. However, the indigenous Ainu from the
northern island of Hokkaido and the Ryukyuans from southern
Okinawa showed <20% Continental specificity, pointing to
them as the most probable descendants of the Jomon people. The
fact that these indigenous groups were, in turn, genetically
well differentiated indicated a notable degree of heterogeneity
and/or isolation among the early Japanese immigrants (Horai et
al. 1996). However, two handicaps of these studies are the
incomplete representation of Asian populations and the
relatively small sample size of those analyzed, which weakens
the reliance on the relative affinities found by genetic
distance methods (Helgason et al. 2001). For mtDNA there are currently enough HVI/HVII
data from eastern Asia, including Japan, to test the validity
of the above-mentioned results. However, these sequences have
been assorted into different clades following different
insufficient criteria or even have not been classified at all.
Furthermore, the phylogenetic confidence of results based only
on sequences from the noncoding region (HVI, HVII) has been
recently questioned (Bandelt et al. 2000). This is mainly due to the frequent occurrence
of parallel mutations in independent lineages that confuse the
correct classification, a source of error that is increased
because the basal motif in the noncoding region for the two
macrolineages that expanded throughout Asia is the same
(16223). In addition, as the noncoding region has not evolved
at a constant rate across all human lineages, it is considered
inappropriate to use this region for dating evolutionary events
(Ingman et al. 2000; Finnilä et al. 2001).
To make reliable use of this important source of available data
on the mtDNA noncoding region to contrast the maternal
structure and to determine the most probable origin of the
modern Japanese, we have undertaken the following approach:
First, we used a set of complete mtDNA sequences of 672
Japanese individuals to create a phylogenetic network (Bandelt
et al. 1999) that related them to other complete sequences,
already published, belonging to the major haplogroups proposed
by others (Torroni et al. 1992, 1996; Macaulay et al. 1999; Yao et al. 2002a). Discriminative positions in the noncoding
region, defining additional Asian subhaplogroups, were then
used to further classify 766 previously published Japanese
partial sequences. For this purpose we also included other
unambiguously assorted sequence data reported by other research
groups (Derbeneva et al. 2002b; Yao et al. 2002a). These HVI sequences thus pooled were then compared
with other published Asian sequences. Finally, using all of
these classified sequences, we tested the relative affinities
of modern Japanese and Continental Asians using global distance
methods and phylogeographic approaches framed at different age
levels.
 |
RESULTS |
Eastern
Asia Phylogeny Based on Complete mtDNA Sequences
The
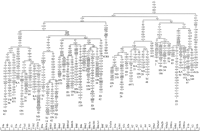
phylogenetic network constructed with the complete mtDNA
sequences fully coincides with those previously published at
worldwide (Maca-Meyer et al. 2001; Herrnstadt et al. 2002) or regional scale (Kong et al. 2003). Moreover, their main branches are well
supported by high bootstrap values on a neighbor-joining tree
(Supplemental material, condensed by more than 40% bootstrap
values).
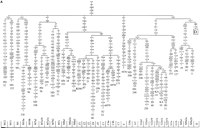
From the L3 African trunk, two early branches came out of Africa
and radiated extensively, originating superhaplogroups M and
N, which were defined by the basic mutations depicted in
Figures 1A and
2,
respectively. Representatives of both superhaplogroups reached
Japan. The construction of these phylogenetic trees by using
our Japanese complete sequences and other published Asian
sequences (Table
1) resulted in a better definition of the known haplogroups
and in the identification of new clades at different
phylogenetic levels. Characteristic HVI motifs and diagnostic
RFLPs in the coding region, and coalescence ages for these
haplogroups and subhaplogroups are given in Supplemental Tables
A and B. To contribute to the unification of the mitochondrial
nomenclature, we revised the previously proposed haplogroups
by adding the following new information.
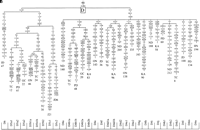
View larger version
(49K):
[in
this window]
[in a new window]
|
Figure 2 Phylogenetic tree, based on complete mtDNA sequences, for
macrohaplogroup N. Origins of subjects are explained in Table
1. The numbers along the links refer to nucleotide
positions, arbitrarily written in ascending order. Open boxes
are nodes from which other (not shown) sequences branch. A, C,
G, and T indicate transversions; whereas "d" indicates
deletions and "i" insertions. Nonrecurrent mutations are
underlined.
Phylogenetic tree, based on complete mtDNA sequences, for
macrohaplogroup N. Origins of subjects are explained in Table
1. The numbers along the links refer to nucleotide
positions, arbitrarily written in ascending order. Open boxes
are nodes from which other (not shown) sequences branch. A, C,
G, and T indicate transversions; whereas "d" indicates
deletions and "i" insertions. Nonrecurrent mutations are
underlined.
| |
Subdivisions
Within Macrohaplogroup M
Haplogroup D
Haplogroup D
has been defined by the specific RFLP -5176 AluI (Torroni et
al. 1992). Studies on Native American HVI sequences
permitted further subdivision of D into subgroups D1 by
mutation 16325 and D2 by mutation 16271 (Forster et al. 1996). Additional subdivisions into subhaplogroups D4
and D5 have been proposed for Asian lineages (Yao et al.
2002a). These investigators characterized D4 by
position 3010. Two additional mutations, 8414 and 14668, have
been proposed to define D4 (Fig.
1B; Kivisild et al. 2002). Whereas these two latter mutations seem to be
rare events, 3010 has also been independently detected in
haplogroups H and J. A new branch at the same phylogenetic
level as D4 and D5 has been detected in Japan (Fig.
1B). It is characterized by mutations 709, 1719, 3714, and
12654 and was named D6. The subdivision of D4 into subgroups
D4a and D4b was proposed on the basis of the distinctive
mutational motif 152, 3206, 14979, and 16129 for the first and
10181 and 16319 for the second (Kivisild et al. 2002). Both subclades have been detected in our Japanese
sample. From our data it can be deduced that mutation 8473 is
also basal for D4a. In relation to D4b it seems that its
ancestral branch is defined by the 8020 substitution (Fig.
1B). Consequently, the D4b subgroup proposed by Yao et al.
(2002a) should be renamed D4b1 harboring 15440 and 15951
as additional basic mutations. A new subgroup characterized by
1382C, 8964, and 9824A mutations and named D4b2, is represented
by lineages GC20 and KA83 in Figure
1B. Furthermore, 12 new branches at the same phylogenetic
level as subhaplogroups D4a and D4b can be identified in the
network. Accordingly, they have been successively named from
D4c to D4n. On the other hand, D5 was defined by mutations 150,
10397, and 16189 (Yao et al. 2002a); however, 16189 is not present in all D5
lineages. We have named D5a and D5b those lineages that share
this mutation and 9180 and D5c those lacking them.
Consequently, we propose to rename D5a of Yao et al. (2002a) as D5a1. Additional mutations (1107 and 5301)
define D5 (Fig.
1B), as has been recently confirmed (Kong et al. 2003). Of the four mutations at the basal branch of
this group, 10397 seems to be a unique event; and the group can
be diagnosed by the RFLP polymorphism +10396 BsrI. Recently,
the phylogeny of haplogroup D has been revised in the light of
complete sequences from Aleuts (Derbeneva et al. 2002b). By comparing their nomenclature to ours, it is
possible to equate their D2 lineage to our D4e1 and their D3
lineage to our D4b1. As a total, D is the most abundant
haplogroup in people of central and eastern Asia including
mainland Japanese but not in the Ainu and Ryukyuans. However,
the geographic distributions of some subhaplogroups are
peculiar. For example, D5 is prevalent in southern areas. D4a
is abundant in Chukchi of northeast Siberia, but D4a1 has its
highest frequency in the Ryukyuans and clade D4n in the Ainu
(Table
2).
Haplogroup
M9
It is confirmed that haplogroup M9 is characterized by
mutation 4491 (Fig.
1A), as recently proposed (Kong et al. 2003). Subhaplogroup M9a, as redefined by Kong et al.
(2003), was identified by positions 153, 3394, 14308,
16234, and 16316 (Yao et al. 2002a). Nevertheless, not all lineages have 153.
Although M9 could be RFLP-diagnosed by +1038 NlaIII and +3391
HaeIII polymorphisms, the latter one should be avoided; as 3391
is also present in some D4d1 lineages (Fig.
1B) and thus could produce misclassification. We have
grouped lineages with 11963 as M9a1 and those with 153 as M9a2.
M9 has a central and eastern Asian geographic distribution,
and it reaches its greatest frequency (11%) and diversity (87%)
in Tibet. In Japan, in addition to mainland Japanese it has
been detected in the indigenous Ainu and Ryukyuans (Horai et
al. 1996).
Haplogroup G
This haplogroup was first detected by Ballinger
et al. (1992) and later named G by Torroni et al. (1994). It was defined by the presence of the combined
RFLP polymorphism +4830 HaeII/+4831 HhaI. In addition, the
basal branch has mutations 709, 5108, and 14569 (Fig.
1; Kivisild et al. 2002). Subhaplogroup G1 was defined by transition
16017 (Schurr et al. 1999) and G2 by mutations 7600 and 16278 (Yao et al.
2002a). Recently, mutations 8200, 15323, and 15497 have
been used for G1 status (Kong et al. 2003). This is confirmed with our Japanese sequences;
consequently, we have defined G1a by 7867 (Fig.
1A). To avoid repetitions, the G1 group of Schurr et al.
(1999) has been provisionally renamed as G5 (Table
2). At least two mutations (5601 and 13563) characterize
G2; and five more, G2a (Fig.
1A; Kong et al. 2003). We have defined subclade G2a1 by the presence
of 16189 and the derivative G2a1a by the addition of 16227,
whereas 16051 and 16150 identify G2a2 lineages. Furthermore,
two new subclades, G3 and G4, are also apparent in Japanese (Fig.
1A). Subgroup G5 is dominant in northeastern Siberia, but
we have not detected it in our set of Japanese complete
sequences. However, G1a1 has its highest frequencies in a
cluster embracing Japanese, Ainu, Ryukyuan, and Koreans. On the
contrary, G2 is relatively abundant in northern China and
central Asia, reaching notable frequencies in the Mansi and in
Tuvinians at the respective west and east ends of South Siberia
(Table
2).
Haplogroup E
Haplogroup E was first RFLP-defined as having
+16389 HinfI and -7598 HhaI by Ballinger et al. (1992), who named it G, and then later it was renamed E
by Torroni et al. (1994). As a loss of restriction sites can be produced
by different nucleotide mutations within the recognition
sequence, since the beginning, some G2 sequences characterized
by the 7600 transition were erroneously classified as belonging
to haplogroup E. Recently, based on the complete sequences of
coding regions, Herrnstadt et al. (2002) defined three Asiatic lineages as E, although only one
(sequence 214) seems to be a genuine representative. It
possesses transition 7598, which, similar to 7600, is also
detectable with HhaI as a site loss; and it also harbors
mutations 10834 and 869, which were found by Ballinger et al.
(1992) as -10830 HinfI and +868 DdeI in all and some
individuals respectively classified as E. However, the
inclusion of a Philippine complete sequence (Ingman and
Gyllensten 2003) in our global tree clearly demonstrates that the
last two mutations might only define a branch of E, as the
Philippine sequence lacks both of them. On the contrary, in
addition to 7598 and 16390, some of the four E mutations
represented in Figure
1A before the branching point might be basic mutations. In
Herrnstadt et al. (2002), sequence 169 belongs to Haplogroup M9 because
it has all coding-region positions defining this haplogroup;
and sequence 287 to M1 because it has 6446 and 6680, the
coding-region mutations that define the basic branch of M1 (Fig.
1). It must be mentioned that the ambiguous Korean lineage
classified as E/G by Schurr et al. (1999), because it had both the -7598 HhaI characteristic E
site and the +4830 HhaI characteristic G site, has been
recently found again in a Korean sample (Snäll et al. 2002). All of them are, in fact, members of
subhaplogroup G2. It seems that haplogroup E has a southern
Asia distribution. Until now it has been detected in the Malay
peninsula populations and in the Sabah of Borneo (Ballinger et
al. 1992); and it is also present in coastal Papua New
Guinea (Stoneking et al. 1990) as well as in some Pacific islands such as Guam
(Herrnstadt et al. 2002) and the Philippines (Ingman and Gyllensten 2003). However, until now, it has not been detected in
more northern Continental populations or islands such as the
Japanese archipelago.
Haplogroup M8
A monophyletic clade (Fig.
1A) groups M8a, C, and Z lineages. Mutations 4715, 15487T,
and 16298 have been proposed as diagnostic for this clade (Yao
et al. 2002a). The transversion 7196A and the transition 8584
should also be included in its definition (Fig.
1A; Kivisild et al. 2002). However, as the 248d is also shared by all Z
and C lineages (Fig.
1A), a basal node defined by this deletion and named CZ has
been recently proposed (Kong et al. 2003). Subhaplogroup C was RFLP-defined by Torroni et
al. (1992) by +13262 AluI. Yao et al. (2002a) added 248d, 14318, and 16327 as characteristic
of C. In addition, positions 3552A, 9545, and 11914 are also
diagnostic of this clade (Fig.
1A; Kivisild et al. 2002). The Japanese TC52 has the C1 status and the
Buryat 6970 and the Evenky 6979 have the C4 status proposed by
Kong et al. (2003). Subhaplogroup Z was defined by Schurr et al.
(1999) by the presence of the following noncoding motifs:
16185, 16223, 16224, 16260, and 16298. Recently, it was
considered that only 16185 and 16260 mutations should be
counted as basic for the group (Yao et al. 2002a). However, in full agreement with the
characterization proposed on the basis of complete Chinese Z
sequences (Kong et al. 2003), three additional mutations (6752, 9090, and
15784) have been placed on the basal branch of Z (Fig.
1A). We detected four Japanese Z clades that, in addition,
shared mutation 152 and another without it. Tentatively, they
have been named from Z1 to Z5 (Fig.
1A). Yao et al. (2002a) defined M8a by 14470, 16184, and 16319
transitions. Two more mutations (6179 and 8684) are also
characteristic of this subhaplogroup (Kong et al. 2003). In Japanese we have found that 16184 is not
harbored by all M8a members. Consequently, lineages with this
mutation have M8a2 status and those lacking it M8a1 status (Fig.
1A). The largest diversities for C are in Korea (100%),
central Asia (86%), and northern China (78%-74%). Therefore,
C can be considered a clade with a Northeast Asian radiation.
Representatives of subhaplogroup Z extend from the Saami
(Finnilä et al. 2001) and Russians (Malyarchuk and Derenko 2001) of west Eurasia to the people of the eastern
peninsula of Kamchatka (Schurr et al. 1999). Its largest diversities are found in Koreans
(88%), northern China (73%), and central Asia (67%), compatible
with a central-East Asian origin of radiation for this group.
Finally, M8a has its highest diversity in Koreans (100%), and
southern (100%) and eastern Chinese, including Taiwanese (73%).
Thus, southeastern China was a potential focus of radiation
of this group. All these subhaplogroups are present in mainland
Japanese but neither in Ryukyuans nor in Ainu.
Haplogroup M7
This haplogroup was defined by Bamshad et al.
(2001) as having two branches, M7a characterized by
16209 and M7b by 16297 transitions. Yao et al. (2002a) assigned mutations 199 and 9824 as basic for M7.
However, our phylogenetic tree points to 6455 and 9824 as the
basal mutations for this group, whereas 199 is only common to
the M7b and M7c subgroups (Fig.
1A), which coincides with the phylogeny proposed by
Kivisild et al. (2002). M7 can be RFLP-diagnosed by the lack of the
6451 MboII restriction site. The M7a subgroup can be defined by
several codingregion positions (Fig.
1A; Kivisild et al. 2002). The M7b classification remains as proposed in
Kivisild et al. (2002); but M7c has, in addition to 146 and 16295,
three more coding-region substitutions (4850, 5442, and 12091)
in its basal branch (Fig.
1A). At this point, it is worthwhile pointing out that the
ambiguously assigned sequence 536 in Herrnstadt et al. (2002) belongs to M7c, as it has the five identifying
coding-region mutations distinctive of this subhaplogroup. As
for the geographic distribution, M7a1 has its highest
frequencies (14%) and diversities (86%) in the Ryukyuans, and
it is also very common in the whole of China, with a mean
diversity of 76%. But, curiously, it has not been detected in
Koreans or in Ainu, and is rare in mainland Japanese. In a
similar way, M7a has its highest diversity in Ryukyuans (83%).
Both groups are rather common in the Philippines. Although M7b
has its greatest diversity in northern China (75%-62%), its
derivative M7b2, has it again in Ryukyuans (100%), Koreans
(53%), and mainland Japanese (45%). On the contrary, M7c is
absent in Ainu and rare in mainland Japanese but very common
in Sabah and the Philippines, although its highest diversity
is in the whole of China (76% ± 11%).
Haplogroup M10
This haplogroup has been defined by
substitutions 10646 and 16311 (Yao et al. 2002a). In addition, Kong et al. (2003) have found several new mutations in its basal
branch that we confirm here (Fig.
1A). Minor modifications are that a new Japanese lineage
shares with M10 only the 8793 mutation, and that a new
mutation, 13152, seems to be basal for our M10 Japanese
lineages. Although its highest frequency is in Tibetans (8%),
the largest diversities are found in China. It is present in
Koreans and mainland Japanese but has not been detected in
either Ainu or Ryukyuans (Table
2).
Haplogroup M11
This haplogroup has been defined by Kong et
al. (2003) by seven coding-region mutations (1095, 6531,
7642, 8108, 9950, 11969, and 13074) and four mutations in
HVS-II (146, 215, 318, and 326). We confirm the same
characterization for our M11 Japanese lineages. A subclade
defined by mutation 14340 was found in Chinese (Kong et al.
2003), but it has not been detected in Japanese. In
turn, Japanese have a new subclade characterized by mutation
14790. Finally, our data suggest that mutation 15924 is at the
root of M11 and the new clade M12.
Haplogroup M12
This haplogroup has been defined in the
present study. It harbors a characteristic motif
(16145-16188-16189-16223-16381) in its noncoding region and
several unique mutations in its coding region (Fig.
1A). Overall, it is a rare haplogroup, being detected only
in mainland Japanese, Koreans, and Tibetans, the lastmentioned
sample showing its highest frequency (8%) and diversity (50%).
Haplogroup M1
Although not present in eastern Asia, this
haplogroup has been included in the phylogenetic tree of
macrohaplogroup M to ascertain its hierarchical level with
respect to other M clades. It was first detected in Ethiopia
(Quintana-Murci et al. 1999) and defined by four transitions in the HVSI
region (16129, 16189, 16249, and 16311). After this, M1 was
also detected in the Mediterranean basin including Jordan
(Maca-Meyer et al. 2001). Several mutations in the coding region are
distinctive of this haplogroup (Fig.
1A). Its RFLP diagnosis is possible by an MnlI site loss
at position 12401.
Subdivisions Within Macrohaplogroup
N
Representatives of two major superhaplogroup N migratory
branches are present in Japan. Two main clades, that directly
sprout from the basal N trunk (A and N9), have a prevailing
northern Asia dispersion, whereas the other two (B and F),
having a southern radiation focus, belong to the derivative R
clade, characterized by the loss of 16223 and 12705 mutations.
Although not detected in Japan, to compare their hierarchical
levels with those of the Asian branches, we have included the
rCRS sequence and a N1b sequence (Kivisild et al. 1999) as representatives of the western Eurasian R and
N clades, respectively.
Haplogroup A
This haplogroup was defined by an HaeIII site
gain at 663 (Torroni et al. 1992). It was subdivided on the basis of HVSI motifs
in A1 (16223-16290-16319) and A2 (16111-16223-16290-16319) by
Forster et al. (1996). In our Japanese sample, we have detected
several A1 representatives characterized by two substitutions
(8563, 11536). Two of these lineages (ON67 and ND218) have been
ascribed to the A1a subgroup that is defined by 4655, 11647,
and 16187 substitutions. Two additional A1 Japanese clusters
(A1b and A1c) have also been phylogenetically defined (Fig.
2). The A2 subgroup is represented in the tree by a
Chukchi (6971) and two (KA21 and ON125) Japanese lineages, all
sharing the 16362 mutation. As the Chukchi harbors the 16111
and 16265 mutations, it has been labeled as an A2a
representative, as tentatively proposed by Saillard et al.
(2000), having four additional mutations (152, 153,
8027, and 12007) in its basal branch. Owing to their
phylogenetic position, three more Japanese lineages (ND28,
TC48, and J42) should be classified as representatives of three
new A subhaplogroups, respectively named A3, A4, and A5 (Fig.
2). Geographically, whereas A1 has a wide northern and
central Asian distribution, subclade A1a is confined to Korea
and mainland Japan. The greatest diversity for A1 is in central
Asia (79%). In Japan it is present in both mainland and
indigenous populations. Subhaplogroup A2 is mainly present in
northeast Siberia including the Kamchatka peninsula, although a
lineage has also been detected in Tibet. The main diversity
(30%) and frequency (60%) for this subhaplogroup are in the
Chukchi.
Subhaplogroups Y, N9a, and N9b
Haplogroup N9 characterized by
the 5417 substitution (Yao et al. 2002a) phylogenetically comprises three subhaplogroups.
Subhaplogroup N9a was mentioned as another N subcluster with
a distinctive HVSI motif (16223, 16257A, 16261) by Richards
et al. (2000). It appears named as N9a in Yao et al. (2002a), who added as basal substitutions 150 and 5231.
Recently, Kong et al. (2003) added mutations 12358 and 12372 at the basal branch
of N9a, which is according to our Japanese phylogeny (Fig.
2). A Japanese N9a1 lineage (TC2) shares mutations 4386,
12007, 16111, and 16129 with the Chinese lineage GD7834 of Kong
et al. (2003). Three more N9a Japanese clusters sharing 16172 as
their basal mutation have been considered distinct N9a2
branches (Fig.
2). Subhaplogroup Y was first identified by a set of HVSI
polymorphisms (16126, 16189, 16231, 16266, 16519), an HaeIII
site loss at 8391 and MboI and DdeI site gains at 7933 and
10394, respectively (Schurr et al. 1999). However, according to the classification of
Kong et al. (2003), all these mutations define the Y1a1 branch
specifically. Our Japanese (Fig.
2) and the Chinese (Kong et al. 2003) phylogenies characterize Y by seven mutations
(8392, 10398, 14178, 14693, 16126, and 16231 gains and a 16223
loss). The branch Y1 would be identified by mutations 3834 and
16266, and the Y1a subcluster by 7933 (Fig.
2; Kong et al. 2003). In Japan we have found a new subclade (Y1b)
characterized by four mutations (146, 10097, 15221, 15460).
Furthermore, a new branch (Y2) with the same phylogenetic
consideration as Y1, and distinguished by six basal mutations
must be aggregated to the Y phylogeny (Fig.
2). Finally, we have detected a sister branch of Y in
Japan. This new lineage, named N9b, shares two basal mutations
(5147 and 16519) with Y and is further characterized by four
(10607, 11016, 13183, 14893) additional mutations in its basal
branch. All N9b1 representatives seem to have the 16189
mutation, and three branches of this trunk (a, b, and c) have
been provisionally defined (Fig.
2). The geographic distribution of subhaplogroup Y is
predominantly in Northeast Asia. The highest frequency (22%) is
in the Ainu, although only one lineage accounts for this
frequency. The greatest diversities are in northern China
(80%), and this group is also very diverse in the Nivkhs from
northeast Siberia (Torroni et al. 1993a). As for N9a, it has a great diversity in the
whole of China (83%) and Korea (79%). In Japan, only mainland
Japanese have N9a representatives. Finally, N9b is very scarce,
being detected in southern China and Korea. Surprisingly, it is
most abundant in the Japanese including the indigenous Ryukyans
and Ainu.
Haplogroup F
This haplogroup was first defined as group A by
Ballinger et al. (1992), and later renamed as F by Torroni et al. (1994). This group was characterized by the lack of
HincII and HpaI sites at 12406. According to the newly proposed
nomenclature (Kivisild et al. 2002; Kong et al. 2003), 12406 is now one of the six mutations that
specifically define subhaplogroup F1. Recently, haplogroup F
has been phylogenetically included as a subcluster of
haplogroup R9 (Yao et al. 2002a). Besides F1, two new subgroups (F2 and F3) have
been defined by Kong et al. (2003). We have found a new subcluster, named F4 (Fig.
2), that is characterized by three coding-region mutations
(5263, 12630, 15670). This group has a particularly high
incidence in Southeast Asia (Ballinger et al. 1992), but only subhaplogroup F1b is well represented
in the Japanese, including the indigenous Ainu and Ryukyuan.
The highest diversities for this subgroup are in eastern China
including Taiwan (100%).
Haplogroup B
Renamed as B after Torroni et al. (1992), this haplogroup was identified by the presence
of a 9-bp deletion in the COII/tRNALys intergenic
region of mtDNA. This polymorphism was first detected in Asia
by RFLP analysis (Cann and Wilson 1983). It was used to classify Japanese on the basis
of the presence/absence of this deletion (Horai and Matsunaga
1986). Even in Asia, the monophyletic status of this
cluster has been repeatedly questioned (Ballinger et al. 1992; Yao et al. 2000b); but although the 9-bp deletion has a high
recurrence, it seems that together with transition 16189 it
defines fairly well a monophyletic cluster, at least in eastern
Asia. Recently, a sister clade of B, keeping the 16189 mutation
but lacking the 9-bp deletion, has been detected in China,
being designated as R11 (Kong et al. 2003). Asian subhaplogroups of B have been named as B4,
identified by the 16217 mutation and B5, characterized by 10398
and 16140 mutations (Yao et al. 2002a). It has been deduced from analysis of complete
sequences that transitions 709, 8584, and 9950 are also in the
basal branch defining B5 (Fig.
2; Kong et al. 2003). Lower-level subdivisions have also been proposed.
Three subclades (B4a, B4b, and B4c) were defined within B4
(Kong et al. 2003). At the same phylogenetic level are our Japanese
branches named B4d, B4e, and B4f; and several new secondary
clusters have also been detected in Japan within B4a, B4b, and
B4c (Fig.
2). It is worthwhile to mention that those lineages
harboring 16189, 16217, 16247, and 16261, also known as the
Polynesian motif (Soodyall et al. 1995), belong to a branch of B4a, having in addition
to 16247, 146, 6719, 12239, 14022, and 15746 as basic
mutations. The B5 cluster was also subdivided in B5a and B5b on
the basis of the HVSI mutations 16266A and 16243, respectively
(Yao et al. 2002a), and reinforced with several additional
positions after the analysis of complete Chinese (Kong et al.
2003) and Japanese (Fig.
2) sequences. Within B5b, new subdivisions are necessary to
accurately classify the Japanese sequences (Fig.
2). Finally, on the basis of characteristic HVSI motifs, we
had tentatively defined as B4a3 those lineages with 16189,
16217, 16261, and 16292 transitions. However, the phylogenetic
position of a Chinese complete sequence (GD7812) belonging to
this HVSI group (Kong et al. 2003) shows that a future redefinition of B4a might be
necessary. The geographic distribution of haplogroup B is very
complex. As expected from its age, the ancestral motif is
widely distributed in Asia excluding Koryacks and other
Siberians. The likewise old subhaplogroup B4 has mainly a
central-eastern Asian distribution with diversities near 100%
from central Asia to Japan. B4a shows a similar distribution as
B4, having branches prevalent in Ryukyuans, Lahu of Yunnan, and
aborigine Taiwanese (Table
2). In a similar vein, some branches of B4c are more
abundant in southern areas (B4c2), whereas others (B4c1) are
mainly detected in Korea and Japan, with derivatives in Taiwan
(B4c1b). On the other hand, subhaplogroup B5a has its greatest
diversity in southern-eastern China (89%), including Taiwan
aborigines (67%), but its B5a1 derivative shows the greatest
diversity in northern China (71%), being present in mainland
Japanese. In turn, subhaplogroup B5b has its major diversity
in Korea (83%) and also reached the Philippines (50%).
Curiously, the B5b1 derivative shows its highest diversity
(67%) and frequency (1%) in mainland Japanese.
Lineage Sorting and Population Pooling
A total of
110 clades with different phylogenetic range have been proposed
on the basis of the pool of the eastern Asian complete
sequences (Figs. 1A,B
and 2). Of
these subdivisions (Table
2), 83 have been used to classify all Asian partial sequences
analyzed in this study. As a test of accuracy in the sorting
of partial sequences into haplogroups, we classified our 672
Japanese complete sequences by using only their HVSI motifs
and found that 34 of them (5%) had an ambiguous status or were
misclassified. The main sources of errors were those sequences
that differed from CRS in only one or two mutations. For
instance, the 16223 mutation was found in M and N backgrounds.
The 16189, 16223 motif can be D6 or N9b. Within M, sorting into
D or G was one of the main sources of ambiguity. Some 16223,
16325, 16362 lineages were D4 and some G1. The motif 16114A,
16223, 16362, classified as D4, was in reality G3. Sometimes
further subdivision within a haplogroup is rather difficult;
for example, there are 16189, 16223, 16362 representatives in
D4 and in D5. Because of recurrency and isolation, it can be
expected that this uncertainty level increases with geographic
distance. For instance, we have found that several 16129, 16223
Japanese lineages belong to D4, but to infer from this that
southern Asian sequences with the same HVSI motif are also D4
would be inappropriate. From a total of 4713 sequences
analyzed, 9.2% had an ambiguous status. In spite of this
percentage there are enough sequences left to carry out
population analysis with statistical confidence.
In a first approach, Japanese, Ainu, and Ryukyuan samples were
compared with the rest of Asian samples shown in Table
3 by means of FST. The closest affinities of
mainland Japanese were to three population groups. The first
include Korean and Han from Shandong (mean P-value =
0.29 ± 0.06), the second Han from Liaoning and Xinjiang, and
the Tu ethnic minority (0.20 ± 0.06), and the third Han from
Xi'an and the Sali, a branch of the Yi ethnic group (0.15 ±
0.06). Ryukyuans and Ainu behave as outliers with significant
differences with all the samples. Population groups resulting
from the FST and CLUSTER analysis are defined in Table
3. Although mainland Japanese from Aichi were significantly
different from other mainland Japanese because of their high
frequency of haplogroup B, they were merged with them as JPN
for comparisons with other areas. Control of the conglomerate
number expected in CLUSTER analysis allows for a hierarchical
grouping of populations. With two conglomerates, the first
distinguished isolate was the aboriginal Sakai from Thailand
(Fucharoen et al. 2001). This group was unique among other Thai people
owing to its lack of lineages with the 9-bp deletion that
characterizes haplogroup B, and to the high frequency of the
authors' C6 cluster (included in our D4a). The lack of any
representative of macrohaplogroup N in a population
anthropologically considered one of the oldest groups in
Thailand, if not caused by genetic drift, is compatible with
the hypothesis that derivatives of macrohaplogroup N had, in
southern Asia, a different route from macrohaplogroup M (Maca-Meyer
et al. 2001). Also striking is the presence in Sakai of an
unequivocal representative (16223-16274-16278-1629416309) of
the sub-Saharan African L2a haplogroup (Torroni et al. 2001), which again is compatible with the physical
characteristics of this Negrito group. Although the suggestion
that the first spreading out of Africa of modern humans could
have carried some L2 lineages in addition to the L3 ancestors
(Watson et al. 1997) is a tempting explanation, a recent admixture is
more in consonance with the phylogenetic proximity of this
lineage to the present African ones. The next outsiders were
the majority of the Siberian isolates, which could not be
pooled because of big differences in the frequency of
distinctive haplogroups (Table
2). This considerable differentiation was already
emphasized (Schurr et al. 1999), with strong genetic drift being its most
probable cause. Subsequent isolates belong to some Chinese
minorities such as those of Lisu and Nu, Lahu, and Taiwanese
aborigines. Unexpectedly, other Chinese minorities (Bai, Sali,
and Tu) were left in Han Chinese northern clusters. The Bai
belong to the Sino-Tibetan Tibeto-Burman ethnic linguistic
group and have been strongly influenced by Han. The Sali are a
minority within the Yi ethnic group whose most probable
ancestors were the Qiang from northwest China. Finally, the Tu,
although belonging to the Mongolian branch of the Altaic
Family, show their main genetic affinities to the Han from
Xi'an (P = 0.95), Xinjiang (P = 0.89), and Shanghai
(P = 0.79), all of them clustered in the Ch2 group. On the
other hand, Thais, Vietnamese, and Cambodians joined with
southern Chinese. As already observed (Chunjie et al. 2000; Yao et al. 2002a), the Han Chinese do not comprise a homogeneous group.
With the exception of cluster Ch4, that includes samples from
Hubei and Guandong (Table
3), they appear geographically differentiated. The two
central Asian groups detected mainly differ in their
frequencies for A1b, Z, and G2a. With less than 14
conglomerates, the Japanese, including Ainu and Ryukyuans, were
part of a big group formed by Korean, Buryat, Tibetans, and
northern Chinese. Ainu was the first differentiated Japanese
sample. Ryukyuans separated later, when mainland Japanese and
Koreans still comprised a single group. The lack of homogeneity
between Ainu and Ryukyuans was pointed out by Horai et al.
(1996), who questioned that they shared a recent common
ancestor. The main differences between them were attributed to
two dominant clusters (C1 and C16, corresponding to our Y and
M5/D4a/G1, respectively) present in Ainu but absent in
Ryukuyans, and two Ryukyuan dominant clusters (C3 and C13,
belonging to our R and M, respectively) absent in Ainu. In
addition, applying the present haplogroup nomenclature to the
same data, the high frequency of M7a1 and D4a1/D4b in
Ryukyuans, but their absence in Ainu, stands out. The MDS plot
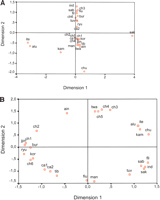
(Fig.
3A), based on FST haplogroup frequency distances
between final groups (data not shown), only partially reflects
the sequential process described above, as only Sakai and
Siberians are well differentiated from the rest. On the
contrary, relationships obtained from haplotype matches (Fig.
3B) show populations highly structured by geography with
the only exceptions being the Ainu and Tuvinian isolates.
The
Peopling of Japan
To further know the relative affinities of
the Japanese between themselves and with the different Asian
groups formed, the data obtained from the global approaches
based on haplogroup frequency distances and on sequence match
identities are presented in Table
4. Both values are moderately correlated in the comparisons
involving the mainland Japanese (r = -0.479; two-tail
probability 0.012) but not at all in those involving aborigine
Ryukyuans (r = -0.310; two-tail probability 0.115) and
Ainu (r = 0.087; two-tail probability 0.667). This
result can be explained by assuming that these aboriginal
people have suffered important genetic drift effects with
substantial changes in haplogroup frequencies and lineage
losses or, less probably, that these populations have been
isolated long enough to have accumulated new variation. Results
based on haplogroup frequencies by far relate mainland Japanese
to Koreans followed by northern Chinese. Ryukyuans present the
smallest distances to Buryats from South Siberia, followed in
short by southern Chinese. In turn, the Ainu have their closest
affinities with mainland Japanese, Koreans, and northern
Chinese. As regards sequence matches, mainland Japanese also
joins first to Koreans and second to Buryats. Aborigine
Ryukyuans are closest to Buryats and then to Koreans. Finally,
Ainu show comparatively less shared sequences, their greater
affinities being toward Chukchi and Koryaks of Kamchatka. This
global picture is congruent with an important influence on
mainland Japanese from northern Asian populations through
Korea, that the Ryukyuans had a dual northern and southern
Asian background previous to the new northern influences
acquired by admixture with mainland Japanese, and that the Ainu
represent the most isolated group in Japan in spite of the
genetic input received from Kamchatka. Also noticeable is the
great distance and low identity values obtained for the
Ainu-Ryukyuan pair compared with those obtained in their
respective comparison to mainland Japanese, which is another
hint of its notable maternal isolation.
View this
table:
[in
this window]
[in a new window]
|
Table 4.
Frequency-Based FST and Sequence Match Identities
(In Percentage) Between Japanese Samples and With Other Asian
Populations
| |
The
distance and identity statistics used above are based on
frequencies of haplogroups and haplotypes, respectively;
however, frequencies are more affected by genetic drift than
the number of different haplotypes present in a population. To
measure the relative affinities of Japanese populations between
them and to Continental Asia in a frequency-independent way, we
chose a haplotype-sharing approach calculating the relative
contribution of lineages shared with other areas to the number
of different haplotypes present in each Japanese population. In
these comparisons all other Asians were merged. Table
5 shows the results of this analysis. Note that despite the
difference in sample size the haplotype frequency in mainland
Japanese and Ainu is 50%, whereas in Ryukyuans it is 84%; which means that,
if there was not a bias in the sampling process, in spite of
its small size, the Ainu sample seems to be representative of
that population. However, it would be desirable to enlarge that
of the Ryukyuans (Helgason et al. 2000). Haplotypes present only in a given population
account for 13% in Ainu but 50% in mainland Japanese
(60%) and Ryukyuans (45%). This finding once more points to the
existence of important drift effects in Ainu. Mainland Japanese
exclusively share with Ryukyuans and Ainu only 3% and 2%,
respectively, of its lineages, which could reach 6% and 3% if
those also shared with Continental Asian populations are added.
In comparison they shared 21% of its lineages with other
Asians. On the contrary, Ryukyuans and Ainu share about 50% of
their lineages with mainland Japanese and only 10% and 21%,
respectively, with Continental populations, which may reflect
other independent Asian influences on Japan. With respect to
those lineages exclusively shared by Japanese and Continental
Asian populations, it is worth mentioning that, again, Korea is
the main contributor, participating in 50% of the haplotype
sharing with mainland Japanese (55%), as much as with Ryukyuans
(50%) and Ainu (50%). However, differences exist in the
provenance of the rest of the shared lineages. Whereas in Ainu
(northern China and Siberia) and in Ryukyuans (northern China
and central Asia) they are from northern areas, the second
region contributing to mainland Japanese is southern China
(17.5%), followed, at the same level (12.5%), by northern China
and central Asia. In addition, there exists a minor percentage
of exclusive sharing with Indonesia (2.5%). On the other hand,
all the matches with Siberia and Tibet are also shared with
other populations. From these results, it can be deduced that
the ancient Japanese inhabitants came from northern Asia and
that southern areas affected the Japanese by later immigration.
Nevertheless, it must be borne in mind that older influences
could be undetectable by lineage sharing. With respect to the
haplogroup affiliation of those lineages that Ainu and
Ryukyuans exclusively shared with no Japanese samples, new
differences appear between them. Ainu share derived lineages of
haplogroups A, G, M9, and D5, all of them compatible with a
rather recent Siberian influence. In contrast, those shared by
Ryukyuans are basical M lineages, more congruent with an older
radiation from southern China. These dual influences are also
detected when the haplogroup affiliation of the Ainu and
Ryukyuan unique lineages is studied. First, the percentage of
lineages belonging to macrohaplogroup N is larger in Ainu (50%)
than in Ryukyuans (15%) and from a different provenance, as
those in Ainu are from haplogroups N, N9b, and Y, whereas those
of Ryukyuans belong to the southern haplogroups F and B. The
remaining 50% of the Ainu lineages equitably belong to
different M haplogroups (M, M7c, G1, and D5a), but in Ryukyuans
the remainder are mainly concentrated in M7a (41%) and M7b2
(18%), two groups that have their greatest Asian diversities
precisely in Ryukyuans. Although an indigenous focus of
radiation cannot be discarded, it is more conservative to
suppose that the most probable origin of these lineages is
again southern China. Thus, Ainu and Ryukyuans are not only
largely isolated populations, but they most probably had
different maternal origins.
Although
no matches are involved, the geographic distribution of
haplogroup frequency and diversities for some groups present in
Japan and in other distinct Asian areas are also relevant to
trace these older connections. For instance, haplogroups M9,
M10, M12, D4b, and F1c have correlated geographic frequencies
with a peak in an area that comprises Tibet (Table
2). Curiously, one of these haplogroups (M12) is today
absent in China but present in Korea and Japan.
|
DISCUSSION |
Although the
recent out-of-Africa origin for all modern humans (Cann et al.
1987) is being widely supported (Takahata et al.
2001), the most probable time and routes chosen by these
earliest migrants to reach eastern Asia is an open issue. In
the following discussion we weigh the different alternatives
proposed in light of the phylogenetic tree obtained from
complete mtDNA sequences. One of the first questions raised was
whether there was more than one out-of-Africa dispersion. All
the mtDNA lineages detected in Old World populations belong to
one of two M and N macrohaplogroups with only secondary
representatives in Africa. The proposed radiation ages for
both, 30,000 to 58,000 years ago and 43,000 to 53,000 years
ago, respectively (Maca-Meyer et al. 2001), give a temporal frame compatible with only one
main dispersion or two successive dispersions, in which case
the M precursor is the most probable candidate for the older
exit. Even if the one dispersion option is chosen, more than
one geographical route to eastern Asia is possible. In fact, a
northern Continental route through the Near East and
western-central Asia and a southern coastal route through the
Arabian and Indian peninsulas have been proposed
(Cavalli-Sforza et al. 1994; Kivisild et al. 1999). The geographical distribution of these two
macrohaplogroups, with lack of ancient M representatives and
the presence of deep N lineages in western Asia, and the
abundance of basal M lineages in India and southwestern Asia
and concomitant lack of equivalent-age N clades, gave rise to
the hypothesis that N represents the main footprint of the
northern Continental expansion, whereas M is the equivalent
footprint for the southern coastal expansion. The presence of N
and M lineages in alternative areas has been explained to have
been the result of secondary migrations (Maca-Meyer et al.
2001). However, another plausible explanation is that
both M and N reached southern Asia at the same time, quickly
expanding to Papua New Guinea (PNG) during maximal glacial ages
when the permafrost boundary precluded a northern human
occupation. During postglacial ages, subsequent migrations
northward carried derivatives of both macrohaplogroups to
northern Asia (Forster et al. 2001). Nevertheless, under this second hypothesis, the
presence of basal N clusters should be expected in India,
southern Asia, and PNG; but this is not the case. All N
representatives in India belong to R, a clade derived from N by
the loss of 16223 and 12705 mutations (Fig.
2). In addition, the bulk of these Indian lineages belong
to western Caucasian haplogroups that, most probably, reached
India as the result of secondary immigrations, as has already
been proposed (Kivisild et al. 1999; Bamshad et al. 2001). Similarly, the N representatives in southern
Asia belong to haplogroups F and B, two sister clades also
derived from R (Fig.
2). Furthermore, when totally sequenced PNG N lineages
(Ingman et al. 2000; Ingman and Gyllensten 2003) are added to the N phylogenetic tree (data not
shown), they form three monophyletic clades that have their
roots in the derived R trunk. On the contrary, the
geographically northern Asian clades A, N9a, N9b, and Y (Fig.
2) and the western Eurasian clades W, N1b, I, and X all
split from the basal N root (Maca-Meyer et al. 2001), although A, N9a, N9b, and Y radiations were delayed
congruent with subsequent northern Asian expansions. Therefore,
at present, mtDNA data are compatible with the supposition that
the northern route, harboring mainly N precursors, met climatic
difficulties and when they finally reached Southeast Asia, the
M representatives, brought by the southern route, had already
colonized the area. This southern expansion of N derivatives
has, as a lower temporal boundary, the coalescence ages of F,
B, and PNG R haplogroups being 46,000 ± 10,000 years
ago. However, when recently published (Ingman et al. 2000; Ingman and Gyllensten 2003) Australian N lineages are taken into account, it
seems evident that the real situation could be far more complex
than the one migration-one lineage hypothesis. Australian N
lineages directly sprout from the basal trunk (data not shown).
They most probably differentiated in that continent, supporting
the idea that ancestral N lineages reached Australia but not
PNG, although the undemonstrable possibility of lineage
extinctions and subsequent recolonization events in PNG can be
an argument. Both hypotheses have difficulties to explain the
presence of ancient N lineages in Australia. If the two, M and
N lineages, were brought with the southern coastal dispersion,
the lack of primitive N in India, southern Asia, and PNG has to
be explained by the subsequent loss of all N lineages carried
to Australia; if the northern Continental route of N is
favored, the loss of N representatives in all populations
formed in route to Australia has also to be explained.
Recently, an N lineage has been detected in Chenchus, a
southern Indian tribal group (Kivisild et al. 2003). From the information published, it can be deduced
that this lineage only shares mutation 1719 with the western
Eurasian Nb1/I and X clades. More extensive studies of
populations in southern India and southern and central Asia
would add empirical support to any of these theories.
Concerning macrohaplogroup M, it has already been commented
that the star radiation of all the main Indian and southeast
Asian M clades strongly suggests that this wide geographic
colonization could have happened in a relatively short time
(Maca-Meyer et al. 2001). This star radiation includes the Australian and PNG
M complete sequences recently published (Ingman et al. 2000; Ingman and Gyllensten 2003). However, for those clades and subclades with
later northward expansions, long radiation delays are observed.
For instance, whereas M7 and M8 have coalescence ages 35,000 to
45,000 years ago, other groups such as G, D4, M7a, or M7c have
coalescence ages 15,000 to 30,000 years ago, more in frame with those
calculated for A, Y, and N9 derivates, which, although
belonging to macrohaplogroup N, share with them a
central-northern Asian geographic distribution (see
Supplemental material). It seems that the simultaneous lineage
bursts 60,000
to 70,000 years ago from Africa (Maca-Meyer et al. 2001), 30,000 to 55,000 years ago for macrohaplogroups M and
N, and 15,000
to 30,000 years ago for clusters with prominent
central-northern Asian radiations were related to main climatic
changes. The role of selection in these expansions is an open
question (Elson et al. 2004; Ruiz-Pesini et al. 2004).
The application of global pairwise-distance and detailed
phylogeographic methods to the peopling of Japan shows that
both approaches have different grasps but together demonstrate
that the actual Japanese population is the result of a complex
demographic history, from which the different theories proposed
to explain it only emphasize partial aspects. Global distances
and detailed haplotype comparisons confirm that Ainu and
Ryukyuans are heterogeneous populations (Horai et al. 1996) and that both are well differentiated from the
mainland Japanese. In spite of this, they have common
peculiarities such as having the highest frequencies in Asia
for M7a, M7b2, and N9b, shared with mainland Japanese.
Furthermore, for both, their closest relatives are northern
populations. At first sight, these results are against a
supposed southern origin for the Paleolithic Japanese, favoring
the replacement theory or even that the Paleolithic inhabitants
of Japan came from northeastern Asia (Nei 1995). Although based on a single locus, our results
are strikingly coincident with the previously proposed northern
origin and influences received by the Japanese. In an early
study using serum gammaglobulin polymorphisms, it was concluded
that the homeland of all Japanese could have been in the Lake
Baikal area in Siberia (Matsumoto 1988), which agrees with the close proximity found
here between Buryats and Ryukyuans or mainland Japanese. More
recently, classical markers (Omoto and Saitou 1997) and mtDNA (Horai et al. 1996) studies demonstrated that the Japanese are most
closely related to the Koreans, which is also true in our
global analysis. It can be added that a substantial part of
this common maternal pool has recent roots, as Korea
specifically shares with Ainu, mainland Japanese, and Ryukyuans
10%, 7%, and 5%, respectively, of their haplotypes. This
particular affinity is increased with the existence of derived
lineages only detected (A1a, B4c1, B4f) or mainly detected
(N9b, B4a1, B4b1, G1a, M7b2, M12) in Japanese and Koreans. This
Korean influence has been attributed to the archeologically
well-documented Continental immigration to Japan during the
Yayoi period (Horai et al. 1996). However, specific haplotype matches with other
areas increases the geographic range of these recent
influences. Thus, mainland Japanese share part of their
haplotypes exclusively with South China (2.5%), North China
(1.5%), Central Asia (1.5%), and Indonesia (0.3%); and, also,
Ryukyuans have specific affinities with North China (2.4%) and
Central Asia (2.4%). The recent Siberian input on the Ainu has
also been stressed (Schurr et al. 1999). At least, another independent migratory wave
from central Asia also affected mainland Japanese. It was first
detected by the peculiar distribution of the Y-chromosome
marker YAP+, and seems to have originated in an area including
Tibet (Su et al. 2000). Haplogroup M12 is its mitochondrial
counterpart. As with the Y-chromosome marker, its punctual
presence in Tibet and eastern Asia might be explained as the
result of subsequent migrations in the Continent that erased
the route followed by the people harboring these markers. In
addition, there are clues, at least in Ryukyuans, that a
substantial part of their maternal pool had an ancient southern
Asian provenance. This fraction is represented by the M, M7a,
and M7a1 basic lineages (31%), which the Ryukyuans do not share
with northern populations. This southern signal is, in part,
congruent with the southern Asian origin for the Paleolithic
Japanese proposed by the dual structure model (Hanihara 1991). Furthermore, the fact that the highest
diversities for M7a, M7a1, and M7b2 have been found in
Ryukyuans and for N9b and B5b2 in Japan raises the possibility
that this area was within a focus of migratory radiations to
northern and southern isles and even to the mainland from
Paleolithic to recent times. The significant latitudinal clines
detected in Japan for some genetic markers (Orito et al. 2001; Takeshita et al. 2001) could also be explained as the result of southern
and northern influences on Japanese. Finally, some mtDNA
results obtained from ancient Jomon remains (Horai et al.
1991; Shinoda and Kanai 1999; K.-I. Shinoda, unpubl.) are congruent with a
genetically diverse background for the Paleolithic Japanese
population (Horai et al. 1996). A tentative comparison of Jomon with
present-day Japanese populations based on shared lineages (data
not shown) significantly relates Jomon first to the indigenous
Ainu and then to Ryukyuans and last to mainland Japanese. In
summary, Japan could have received several northern and
southern Asian maternal inputs since Paleolithic times, with
notable northern Asian immigrations through Korea in the late
Neolithic and more specific gene flows from western Asia,
Siberia, and southern islands.
|
METHODS |
Samples
Complete
mtDNA sequences were obtained from a total of 672 unrelated
Japanese including 373 from Tokyo and 299 from the Nagoya area.
All subjects gave their written consent to participate in this
study, which was approved by the Ethical Committees of the Gifu
International Institute of Biotechnology and collaborative
institutions. The sources of 11 additional complete sequences
used to build the final phylogenetic trees are in Table
1. For the analysis of the peopling of Japan, we used a
total of 1438 Japanese and 3275 central and eastern Asian HVI
sequences, as detailed in Table
3.
Isolation and Amplification of DNA
Total DNA was
extracted from the blood with either Dr. Gen TLE (Takara) or
MagExtractor System MFX-2000 (Toyobo). The entire mitochondrial
genome was amplified as six fragments (3000-3400 bp)
by the first PCR and 60 overlapping segments (600-1000 bp) by
the second PCR. The primer pairs and their nucleotide sequences
were described previously (Tanaka et al. 1996). The conditions for the first and second PCR
were the same: an initial denaturation step for 5 min at 94°C,
followed by 40 cycles of denaturation for 15 sec at 94°C,
annealing for 15 sec at 60°C, and extension for 3 min at 72°C,
with a final extension for 10 min at 72°C. The amplified
fragments were analyzed by electrophoresis on a 1% agarose gel
and visualized by staining with ethidium bromide. These second
PCR products were purified by use of the MultiScreen-PCR Plates
(Millipore). The quality of DNA templates was examined by
electrophoresis on a 1.2% agarose gel after staining with
ethidium bromide by use of a Ready-To-Run Separation Unit
(Amersham Pharmacia Biotech).
Sequence Analysis of Mitochondrial DNA
Sequence
reactions were carried out with a BigDye terminator cycle
sequencing FS ready reaction kit (Applied Biosystems). After
excess dye terminators had been removed with MultiScreen-HV
plates (Millipore) packed with Sephadex G50 superfine
(Pharmacia), the purified DNA samples were precipitated with
ethanol, dried, and suspended in the template suppression
reagent (TSR) or formamide from Applied Biosystems. The
dissolved DNA samples were heated for 2 min at 95°C for
denaturation, then immediately cooled on ice. Sequences were
analyzed with automated DNA sequencers 377 and 310 by use of
Sequencing Analysis Program version 4.1 (Applied Biosystems). A
computer program, Sequencher version 4.1 (Gene Codes Co.), was
used to indicate possible single nucleotide polymorphism (SNP)
loci. For verification, visual inspection of each candidate SNP
was carried out. At least two overlapping DNA templates
amplified with different primer pairs were used for
identification of each SNP. Mitochondrial SNPs (mtSNPs) were
identified by comparison with the revised Cambridge sequence
(rCRS) reported by Andrews et al. (1999).
Phylogenetic Analysis of Complete Coding-Region mtDNA
Sequences
In this present study, nucleotide positions were
numbered as in the Cambridge Reference Sequence (CRS; Anderson
et al. 1981), nucleotide substitutions were expressed as
differences from the revised CRS (Andrews et al. 1999), transitions were denoted only by their
nucleotide positions, and transversions were designated by
their nucleotide positions followed by the changed base. A
total of 942 complete coding-region mtDNA sequences, including
our 672 Japanese; one additional Japanese (GenBank accession
no. AB055387
[GenBank]
); 53 worldwide sequences (Ingman et al. 2000); 42 worldwide sequences (Maca-Meyer et al.
2001); two Finnish sequences having Asian relatives
(Finnilä et al. 2001); 17 Asian sequences without concrete geographic
assignation (Herrnstadt et al. 2002); 37 sequences from the Bering area (Derbeneva et
al. 2002b); 70 Asian, New Guinean, and Australian sequences
(Ingman and Gyllensten 2003); and 48 Chinese sequences (Kong et al. 2003) were aligned with the rCRS by CLUSTAL V software,
and the coding region was used to construct a phylogenetic
network (Bandelt et al. 1999) rooted with a chimpanzee sequence (GenBank
accession no. D38113
[GenBank]
) as implemented in the Network 3.1 program
(Fluxus Engineering; http://www.fluxus-engineering.com/).
The noncoding positions were added by hand using molecular
weighted parsimony criteria (Bandelt et al. 2000). The phylogenetic relationships obtained were
also confirmed by means of a neighbor-joining tree (1000x bootstrapped; Saitou and Nei 1987), built using MEGA2 (Kumar et al. 2001). From this network (see Supplemental material)
we chose 102 Japanese and nine Asiatic sequences that
represented the main clusters and subclusters within the two
macrohaplogroups M and N that colonized Asia. To define these
groups we followed the most generalized cladistic nomenclature
actually used to classify mtDNA lineages (Richards et al. 1998). For the haplogroups previously detected, we
maintained the same notation as their authors proposed
(Richards et al. 2000; Bamshad et al. 2001; Kivisild et al. 2002; Yao et al. 2002a; Kong et al. 2003). Those haplogroups introduced here for the first time
were named according to their phylogenetic range deduced from
the tree of complete sequences.
Haplogroup Assorting of Published Partial mtDNA
Sequences
The unambiguously classified complete mtDNA
sequences were used as an initial pool that was hierarchically
enlarged by the successive addition of those published partial
mtDNA sequences with the largest coding information, ending
with those for which information on only control-region
sequences for both mtDNA hypervariable segments or just one
(HVS-I and/or HVS-II) was available, always following sequence
matches or, as default, sequence-relatedness criteria. Some of
those partial sequences that could be assigned to more than one
haplogroup were tentatively assorted in the most probable one
deduced from their geographic origin and the relative
haplogroup distribution.
Pooling Small Size Samples and Rare Clades
To avoid
small sample sizes and rare alleles in population comparisons,
samples with <20 individuals were pooled with others from
the same geographic and ethnic group. Within populations,
individuals belonging to rare clades were pooled with those
classified in the nearest branch. Pairwise sample distances
were calculated as linearized FST distances as
implemented in the ARLEQUIN program (Schneider et al. 2000), taking mtDNA as one locus with as many alleles
as the different subhaplogroups considered.
Quantitative Affinities of Japanese
Samples
Relative affinities of Japanese samples to the other
Asiatic populations were assessed by linearized FST
distances, using subhaplogroup frequencies, and haplotype
matches' distances (D) estimated simply as D = 1
- ![{sum}]() (xiyi),
xi and yi being the
frequency of haplotype i in the two compared populations.
To be statistically robust, these analyses require large
sample sizes, thus further pooling was necessary. Previous
studies in the area prevented us from pooling populations by
geographic proximity (Schurr et al. 1999) and/or ethno-linguistic relationship (Comas et
al. 1998; Chunjie et al. 2000; Yao et al. 2002a). For this reason, a genetic affinity criterion
was chosen. Two approaches were used. In the first, all samples
with no significant FST distances between them and
with a similar behavior to the rest of the samples studied,
were grouped. In the second, pooling was carried out by means
of the CLUSTER algorithm implemented in the SPSS ver 9 package.
We followed an iterative method specifying the number of
conglomerates from 2 to 30. Different groupings were tested by
AMOVA, and that with the least assigned variance within areas
was chosen. The data were graphically represented by
multidimensional scaling (MDS) plots (Kruskal and Wish 1978) using SPSS. (xiyi),
xi and yi being the
frequency of haplotype i in the two compared populations.
To be statistically robust, these analyses require large
sample sizes, thus further pooling was necessary. Previous
studies in the area prevented us from pooling populations by
geographic proximity (Schurr et al. 1999) and/or ethno-linguistic relationship (Comas et
al. 1998; Chunjie et al. 2000; Yao et al. 2002a). For this reason, a genetic affinity criterion
was chosen. Two approaches were used. In the first, all samples
with no significant FST distances between them and
with a similar behavior to the rest of the samples studied,
were grouped. In the second, pooling was carried out by means
of the CLUSTER algorithm implemented in the SPSS ver 9 package.
We followed an iterative method specifying the number of
conglomerates from 2 to 30. Different groupings were tested by
AMOVA, and that with the least assigned variance within areas
was chosen. The data were graphically represented by
multidimensional scaling (MDS) plots (Kruskal and Wish 1978) using SPSS.
Qualitative Affinities of Japanese
Samples
Particular sharing of subhaplogroups and particular
haplotype matches of Japanese samples with concrete Continental
areas were phylogeographically analyzed by taking into account
the relative genetic diversities of the clades involved in the
different areas, measured as relative haplotypic frequencies,
and their minimum estimates of coalescence ages based on mean
divergence among lineages for the coding region (Saillard et
al. 2000). A constant evolutionary rate of 1.7 x 10-8 per site per year
(Ingman et al. 2000) was used.
|
Acknowledgements |
This work was
supported in part by the Support Project for Database
Development from the Japan Science and Technology Corporation
(to M.T.), Grants-in-Aid for Scientific Research (C2-10832009,
A2-15200051) and for Priority Areas from the Ministry of
Education, Science, Sports and Culture of Japan (to M.T.), and
by grants BMC2001-3511 and COF2002-015 (to V.M.C.).
|
Footnotes |
15 Corresponding author.
E-MAIL mtanaka{at}giib.or.jp
; FAX 81-583-71-4412. ![Back]()
[Supplemental material is available online
at www.genome.org.]
Article and publication are at
http://www.genome.org/cgi/doi/10.1101/gr.2286304.
|
REFERENCES |
Abe, S., Usami, S., Shinkawa, H., Weston,
M.D., Overbeck, L.D., Hoover, D.M., Kenyon, J.B., Horai, S., and
Kimberling, W.J. 1998. Phylogenetic analysis of mitochondrial DNA in
Japanese pedigrees of sensorineural hearing loss associated with the
A1555G mutation. Eur. J. Hum. Genet. 6:
563-569.[CrossRef][Medline]
Anderson, S., Bankier, A.T., Barrell,
B.G., de Bruijn, M.H., Coulson, A.R., Drouin, J., Eperon, I.C., Nierlich,
D.P., Roe, B.A., Sanger, F., et al. 1981. Sequence and organization of the
human mitochondrial genome. Nature 290:
457-465.[CrossRef][Medline]
Andrews, R.M., Kubacka, I., Chinnery,
P.F., Lightowlers, R.N., Turnbull, D.M., and Howell, N. 1999. Reanalysis
and revision of the Cambridge reference sequence for human mitochondrial
DNA. Nat. Genet. 23: 147.[CrossRef][Medline]
Ballinger, S.W., Schurr, T.G., Torroni,
A., Gan, Y.Y., Hodge, J.A., Hassan, K., Chen, K.H., and Wallace, D.C.
1992. Southeast Asian mitochondrial DNA analysis reveals genetic
continuity of ancient mongoloid migrations. Genetics
130:
139-152.[Abstract]
Bamshad, M., Kivisild, T., Watkins, W.S.,
Dixon, M.E., Ricker, C.E., Rao, B.B., Naidu, J.M., Prasad, B.V., Reddy,
P.G., Rasanayagam, A., et al. 2001. Genetic evidence on the origins of
Indian caste populations. Genome Res. 11:
994-1004.[Abstract/Free Full Text]
Bandelt, H.-J., Forster, P., and Röhl, A.
1999. Median-joining networks for inferring intraspecific phylogenies.
Mol. Biol. Evol. 16: 37-48.[Abstract]
Bandelt, H.-J., Macaulay, V., and
Richards, M. 2000. Median networks: Speedy construction and greedy
reduction, one simulation, and two case studies from human mtDNA. Mol.
Phylogenet. Evol. 16: 8-28.[CrossRef][Medline]
Betty, D.J., Chin-Atkins, A.N., Croft, L.,
Sraml, M., and Easteal, S. 1996. Multiple independent origins of the
COII/tRNA(Lys) intergenic 9-bp mtDNA deletion in aboriginal Australians.
Am. J. Hum. Genet. 58: 428-433.[Medline]
Cann, R.L. and Wilson, A.C. 1983. Length
mutations in human mitochondrial DNA. Genetics
104: 669-711.
Cann, R.L., Stoneking, M., and Wilson,
A.C. 1987. Mitochondrial DNA and human evolution. Nature
325: 31-36.[CrossRef][Medline]
Cavalli-Sforza, L.L., Menozzi, P., and
Piazza, A. 1994. The history and geography of human genes.
Princeton University Press, Princeton, NJ.
Chunjie, X., Cavalli-Sforza, L.L.,
Minch, E., and Ruofu, D.U. 2000. Principal component analysis of gene
frequencies of Chinese populations. Science in China Ser. C
43: 472-481.
Comas, D., Calafell, F., Mateu, E.,
Perez-Lezaun, A., Bosch, E., Martinez-Arias, R., Clarimon, J., Facchini,
F., Fiori, G., Luiselli, D., et al. 1998. Trading genes along the silk
road: mtDNA sequences and the origin of central Asian populations. Am.
J. Hum. Genet. 63: 1824-1838.[CrossRef][Medline]
Derbeneva, O.A., Starikovskaya, E.B.,
Wallace, D.C., and Sukernik, R.I. 2002a. Traces of early Eurasians in the
Mansi of northwest Siberia revealed by mitochondrial DNA analysis. Am.
J. Hum. Genet. 70: 1009-1014.[CrossRef][Medline]
Derbeneva, O.A., Sukernik, R.I.,
Volodko, N.V., Hosseini, S.H., Lott, M.T., and Wallace, D.C. 2002b.
Analysis of mitochondrial DNA diversity in the Aleuts of the commander
islands and its implications for the genetic history of Beringia. Am.
J. Hum. Genet. 71: 415-421.[CrossRef][Medline]
Derenko, M.V., Malyarchuk, B.A., Dambueva,
I.K., Shaikhaev, G.O., Dorzhu, C.M., Nimaev, D.D., and Zakharov, I.A.
2000. Mitochondrial DNA variation in two South Siberian Aboriginal
populations: Implications for the genetic history of North Asia. Hum.
Biol. 72: 945-973.[Medline]
Elson, J.L., Turnbull, D.M., and Howell,
N. 2004. Comparative genomics and the evolution of human mitochondrial
DNA: Assessing the effects of selection. Am. J. Hum. Genet.
74:
229-238.[CrossRef][Medline]
Finnilä, S., Lehtonen, M.S., and
Majamaa, K. 2001. Phylogenetic network for European mtDNA. Am. J. Hum.
Genet. 68: 1475-1484.[CrossRef][Medline]
Forster, P., Harding, R., Torroni, A.,
and Bandelt, H.J. 1996. Origin and evolution of Native American mtDNA
variation: A reappraisal. Am. J. Hum. Genet.
59:
935-945.[Medline]
Forster, P., Torroni, A., Renfrew, C.,
and Röhl, A. 2001. Phylogenetic star contraction applied to Asian and
Papuan mtDNA evolution. Mol. Biol. Evol.
18: 1864-1881.[Abstract/Free Full Text]
Fucharoen, G., Fucharoen, S., and Horai,
S. 2001. Mitochondrial DNA polymorphisms in Thailand. J. Hum.
Genet. 46: 115-125.[CrossRef][Medline]
Glover, I.C. 1980. Agricultural origins
in East Asia. In The Cambridge encyclopedia of archaeology (ed. A.
Sherratt), pp. 152-161. Crown, New York.
Hammer, M.F. and Horai, S. 1995. Y
chromosomal DNA variation and the peopling of Japan. Am. J. Hum.
Genet. 56: 951-962.[Medline]
Hanihara, K. 1991. Dual structure model
for the population history of the Japanese. Japan Review
2: 1-33.
Helgason, A., Sigureth Ardottir, S.,
Gulcher, J.R., Ward, R., and Stefansson, K. 2000. mtDNA and the origin of
the Icelanders: Deciphering signals of recent population history. Am.
J. Hum. Genet. 66: 999-1016.[CrossRef][Medline]
Helgason, A., Hickey, E., Goodacre, S.,
Bosnes, V., Stefánsson, K., Ward, R., and Sykes, B. 2001. mtDNA and the
islands of the North Atlantic: Estimating the proportions of Norse and
Gaelic ancestry. Am. J. Hum. Genet. 68:
723-737.[CrossRef][Medline]
Herrnstadt, C., Elson, J.L., Fahy, E.,
Preston, G., Turnbull, D.M., Anderson, C., Ghosh, S.S., Olefsky, J.M.,
Beal, M.F., Davis, R.E., et al. 2002. Reduced-median-network analysis of
complete mitochondrial DNA coding-region sequences for the major African,
Asian, and European haplogroups. Am. J. Hum. Genet.
70: 1152-1171.[CrossRef][Medline]
Horai, S. and Hayasaka, K. 1990.
Intraspecific nucleotide sequence differences in the major noncoding
region of human mitochondrial DNA. Am. J. Hum. Genet.
46:
828-842.[Medline]
Horai, S. and Matsunaga, E. 1986.
Mitochondrial DNA polymorphism in Japanese. II. Analysis with restriction
enzymes of four or five base pair recognition. Hum. Genet.
72:
105-117.[Medline]
Horai, S., Kondo, R., Murayama, K.,
Hayashi, S., Koike, H., and Nakai, N. 1991. Phylogenetic affiliation of
ancient and contemporary humans inferred from mitochondrial DNA. Phil.
Trans. R Soc. Lond. B 333: 409-417.[CrossRef][Medline]
Horai, S., Murayama, K., Hayasaka, K.,
Matsubayashi, S., Hattori, Y., Fucharoen, G., Harihara, S., Park, K.S.,
Omoto, K., and Pan, I.H. 1996. mtDNA polymorphism in East Asian
Populations, with special reference to the peopling of Japan. Am. J.
Hum. Genet. 59: 579-590.[Medline]
Imaizumi, K., Parsons, T.J., Yoshino, M.,
and Holland, M.M. 2002. A new database of mitochondrial DNA hypervariable
regions I and II sequences from 162 Japanese individuals. Int. J.
Legal. Med. 116: 68-73.[CrossRef][Medline]
Ingman, M. and Gyllensten, U. 2003.
Mitochondrial genome variation and evolutionary history of Australian and
New Guinean Aborigines. Genome Res. 13:
1600-1606.[Abstract/Free Full Text]
Ingman, M., Kaessmann, H., Pääbo, S.,
and Gyllensten, U. 2000. Mitochondrial genome variation and the origin of
modern humans. Nature 408: 708-713.[CrossRef][Medline]
Jorde, L.B., Bamshad, M.J., Watkins, W.S.,
Zenger, R., Fraley, A.E., Krakowiak, P.A., Carpenter, K.D., Soodyall, H.,
Jenkins, T., and Rogers, A.R. 1995. Origins and affinities of modern
humans: A comparison of mitochondrial and nuclear genetic data. Am. J.
Hum. Genet. 57: 523-538.[Medline]
Kivisild, T., Kaldma, K., Metspalu, M.,
Parik, J., Papiha, S., and Villems, R. 1999. The place of the Indian
mitochondrial DNA variants in the global network of maternal lineages and
the peopling of the Old World. In Genomic diversity: Applications in
human population genetics (eds. S. Papiha et al.), pp. 135-152. Plenum
Press, New York.
Kivisild, T., Tolk, H.-V., Parik, J.,
Wang, Y., Papiha, S.S., Bandelt, H.-J., and Villems, R. 2002. The emerging
limbs and twigs of the East Asian mtDNA tree. Mol. Biol. Evol.
19: 1737-1751.[Abstract/Free Full Text]
Kivisild, T., Rootsi, S., Metspalu, M.,
Mastana, S., Kaldma, K., Parik, J., Metspalu, E., Adojaan, M., Tolk, H.V.,
Stepanov, V., et al. 2003. The genetic heritage of the earliest settlers
persists both in Indian tribal and caste populations. Am. J. Hum.
Genet. 72: 313-332.[CrossRef][Medline]
Kolman, C.J., Sambuughin, N., and
Bermingham, E. 1996. Mitochondrial DNA analysis of Mongolian populations
and implications for the origin of New World founders. Genetics
142: 1321-1334.[Abstract]
Kong, Q.-P., Yao, Y.-G., Sun, C.,
Bandelt, H.-J., Zhu, C.-L., and Zhang, Y.-P. 2003. Phylogeny of East Asian
mitochondrial DNA lineages inferred from complete sequences. Am. J.
Hum. Genet. 73: 671-676.[CrossRef][Medline]
Koyama, H., Iwasa, M., Maeno, Y.,
Tsuchimochi, T., Isobe, I., Seko-Nakamura, Y., Monma-Ohtaki, J.,
Matsumoto, T., Ogawa, S., Sato, B., et al. 2002. Mitochondrial sequence
haplotype in the Japanese population. Forensic Sci. Int.
125: 93-96.[CrossRef][Medline]
Kruskal, J.B. and Wish, M. 1978.
Multidimensional scaling. Sage Publications, Beverly Hills, CA.
Kumar, S., Tamura, K., Jakobsen, I.B.,
and Nei, M. 2001. MEGA2: Molecular Evolutionary Genetics Analysis
software. Bioinformatics 17: 1244-1245.[Abstract/Free Full Text]
Lee, S.D., Shin, C.H., Kim, K.B., Lee,
Y.S., and Lee, J.B. 1997. Sequence variation of mitochondrial DNA control
region in Koreans. Forensic Sci. Int. 87:
99-116.[CrossRef][Medline]
Lee, S.D., Lee, Y.S., and Lee, J.B. 2002.
Polymorphism in the mitochondrial cytochrome B gene in Koreans. An
additional marker for individual identification. Int. J. Legal Med.
116: 74-78.[CrossRef][Medline]
Maca-Meyer, N., González, A.M., Larruga,
J.M., Flores, C., and Cabrera, V.M. 2001. Major genomic mitochondrial
lineages delineate early human expansions. BMC Genet.
2: 13-20.[CrossRef][Medline]
Macaulay, V., Richards, M., Hickey, E.,
Vega, E., Cruciani, F., Guida, V., Scozzari, R., Bonne-Tamir, B., Sykes,
B., and Torroni, A. 1999. The emerging tree of West Eurasian mtDNAs: A
synthesis of control-region sequences and RFLP. Am. J. Hum. Genet.
64:
232-249.[CrossRef][Medline]
Malyarchuk, B.A. and Derenko, M.V. 2001.
Mitochondrial DNA variability in Russians and Ukrainians: Implication to
the origin of the Eastern Slavs. Ann. Hum. Genet.
65: 63-78.[CrossRef][Medline]
Matsumoto, H. 1988. Characteristics of
Mongoloid and neighboring populations based on the genetic markers of
human immunoglobulins. Hum. Genet. 80:
207-218.[CrossRef][Medline]
Melton, T., Clifford, S., Martinson, J.,
Batzer, M., and Stoneking, M. 1998. Genetic evidence for the
proto-Austronesian homeland in Asia: mtDNA and nuclear DNA variation in
Taiwanese aboriginal tribes. Am. J. Hum. Genet.
63: 1807-1823.[CrossRef][Medline]
Nei, M. 1995. The origins of human
populations: Genetic, linguistic, and archeological data. In The origin
and past of modern humans as viewed from DNA (eds. S. Brenner and K.
Hanihara), pp. 71-91. World Scientific, Singapore.
Nishimaki, Y., Sato, K., Fang, L., Ma, M.,
Hasekura, H., and Boettcher, B. 1999. Sequence polymorphism in the mtDNA
HV1 region in Japanese and Chinese. Legal Med.
1: 238-249.[CrossRef][Medline]
Omoto, K. and Saitou, N. 1997. Genetic
origins of the Japanese: A partial support for the dual structure
hypothesis. Am. J. Phys. Anthropol. 102:
437-446.[CrossRef][Medline]
Oota, H., Kitano, T., Jin, F., Yuasa, I.,
Wang, L., Ueda, S., Saitou, N., and Stoneking, M. 2002. Extreme mtDNA
homogeneity in continental Asian populations. Am. J. Phys.
Anthropol. 118: 146-153.[CrossRef][Medline]
Orito, E., Ichida, T., Sakugawa, H.,
Sata, M., Horiike, N., Hino, K., Okita, K., Okanoue, T., Iino, S., Tanaka,
E., et al. 2001. Geographic distribution of hepatitis B virus (HBV)
genotype in patients with chronic HBV infection in Japan.
Hepatology 34: 590-594.[CrossRef][Medline]
Pfeiffer, H., Steighner, R., Fisher, R.,
Mornstad, H., Yoon, C.L., and Holland, M.M. 1998. Mitochondrial DNA
extraction and typing from isolated dentin-experimental evaluation in a
Korean population. Int. J. Legal Med. 111:
309-313.[CrossRef][Medline]
Qian, Y.P., Chu, Z.T., Dai, Q., Wei,
C.D., Chu, J.Y., Tajima, A., and Horai, S. 2001. Mitochondrial DNA
polymorphisms in Yunnan nationalities in China. J. Hum. Genet.
46:
211-220.[CrossRef][Medline]
Quintana-Murci, L., Semino, O., Bandelt,
H.-J., Passarino, G., McElreavey, K., and Santachiara-Benereccetti, A.S.
1999. Genetic evidence of an early exit of Homo sapiens sapiens
from Africa through eastern Africa. Nat. Genet.
23:
437-441.[CrossRef][Medline]
Redd, A.J. and Stoneking, M. 1999. Peopling
of Sahul: mtDNA variation in aboriginal Australian and Papua New Guinean
populations. Am. J. Hum. Genet. 65:
808-828.[CrossRef][Medline]
Richards, M., Macaulay, V., Bandelt,
H.-J., and Sykes, B. 1998. Phylogeography of mitochondrial DNA in western
Europe. Ann. Hum. Genet. 62: 241-260.[CrossRef][Medline]
Richards, M., Macaulay, V., Hickey, E.,
Vega, E., Sykes, B., Guida, V., Rengo, C., Sellitto, D., Cruciani, F.,
Kivisild, T., et al. 2000. Tracing European founder lineages in the Near
Eastern mtDNA pool. Am. J. Hum. Genet. 67:
1251-1276.[Medline]
Ruiz-Pesini, E., Mishmar, D., Brandon,
M., Procaccio, V., and Wallace, D.C. 2004. Effects of purifying and
adaptive selection on regional variation in human mtDNA. Science
303:
223-226.[Abstract/Free Full Text]
Saillard, J., Forster, P., Lynnerup, N.,
Bandelt, H.J., and Norby, S. 2000. mtDNA variation among Greenland
Eskimos: The edge of the Beringian expansion. Am. J. Hum. Genet.
67:
718-726.[CrossRef][Medline]
Saitou, N. and Nei, M. 1987. The
neighbor-joining method: A new method for reconstructing phylogenetic
trees. Mol. Biol. Evol. 4: 406-425.[Abstract]
Schneider, S., Roessli, D., and
Excoffier, L. 2000. Arlequin ver. 2000: A software for population
genetics data analysis. Genetic and Biometry Laboratory, University of
Geneva, Switzerland.
Schurr, T.G., Sukernik, R.I.,
Starikovskaya, Y.B., and Wallace, D.C. 1999. Mitochondrial DNA variation
in Koryaks and Itel'men: Population replacement in Okhotsk Sea-Bering Sea
region during the Neolithic. Am. J. Phys. Anthropol.
108: 1-39.[Medline]
Seo, Y., Stradmann-Bellinghausen, B.,
Rittner, C., Takahama, K., and Schneider, P.M. 1998. Sequence polymorphism
of mitochondrial DNA control region in Japanese. Forensic Sci. Int.
97:
155-164.[CrossRef][Medline]
Shields, G.F., Schmiechen, A.M., Frazier,
B.L., Redd, A., Voevoda, M.I., Reed, J.K., and Ward, R.H. 1993. mtDNA
sequences suggest a recent evolutionary divergence for Beringian and
northern North American populations. Am. J. Hum. Genet.
53:
549-562.[Medline]
Shinoda, K.-I. and Kanai, S. 1999.
Intracemetry genetic analysis at the Nakazuma Jomon site in Japan by
mitochondrial DNA sequencing. Anthropol. Sci.
107: 129-140.
Shiraishi, T. 2002. Wakoku tanjou (The
formation of ancient Japanese society). In History of Japan 1 (ed.
T. Shiraishi et al.), pp. 8-94. Yoshikawakobunkan, Tokyo, Japan (in
Japanese).
Snäll, N., Savontaus, M.-L., Kares, S.,
Lee, M.S., Cho, E.K., Rinne, J.O., and Huoponen, K. 2002. A rare
mitochondrial DNA haplotype observed in Koreans. Hum. Biol.
74:
253-262.[Medline]
Soodyall, H., Jenkins, T., and
Stoneking, M. 1995. `Polynesian' mtDNA in the Malagasy. Nat. Genet.
10:
377-378.[CrossRef][Medline]
Stoneking, M., Jorde, L.B., Bhatia, K.,
and Wilson, A.C. 1990. Geographic variation in human mitochondrial DNA
from Papua New Guinea. Genetics 124:
717-733.[Abstract]
Su, B., Xiao, C., Deka, R., Seielstad,
M.T., Kangwanpong, D., Xiao, J., Lu, D., Underhill, P., Cavalli-Sforza,
L., Chakraborty, R., et al. 2000. Y chromosome haplotypes reveal
prehistorical migrations to the Himalayas. Hum. Genet.
107:
582-590.[CrossRef][Medline]
Sykes, B., Leiboff, A., Low-Beer, J.,
Tetzner, S., and Richards, M. 1995. The origins of the Polynesians: An
interpretation from mitochondrial lineage analysis. Am. J. Hum.
Genet. 57: 1463-1475.[Medline]
Tajima, A., Sun, C.-S., Pan, I.-H., Ishida,
T., Saitou, N., and Horai, S. 2003. Mitochondrial DNA polymorphisms in
nine aboriginal groups of Taiwan: Implications for the population history
of aboriginal Taiwanese. Hum. Genet. 113:
24-33.[CrossRef][Medline]
Takahata, N., Lee, S.-H., and Satta, Y.
2001. Testing multiregionality of modern human origins. Mol. Biol.
Evol. 18: 172-183.[Abstract/Free Full Text]
Takeshita, T., Yasuda, Y., Nakashima,
K., Mogi, K., Kishi, H., Shiono, K., Sagisaka, I., Yuasa, H., Nishimukai,
H., and Kimura, H. 2001. Geographical north-south decline in
DNASE*2 in Japanese populations. Hum. Biol.
73:
129-134.[Medline]
Tanaka, M., Hayakawa, M., and Ozawa, T.
1996. Automated sequencing of mitochondrial DNA. Methods Enzymol.
264:
407-421.[Medline]
Torroni, A., Schurr, T.G., Yang, C.C.,
Szathmary, E.J., Williams, R.C., Schanfield, M.S., Troup, G.A., Knowler,
W.C., Lawrence, D.N., Weiss, K.M., et al. 1992. Native American
mitochondrial DNA analysis indicates that the Amerind and the Nadene
populations were founded by two independent migrations. Genetics
130:
153-162.[Abstract]
Torroni, A., Sukernik, R.I., Schurr,
T.G., Starikorskaya, Y.B., Cabell, M.F., Crawford, M.H., Comuzzie, A.G.,
and Wallace, D.C. 1993a. mtDNA variation of aboriginal Siberians reveals
distinct genetic affinities with Native Americans. Am. J. Hum.
Genet. 53: 591-608.[Medline]
Torroni, A., Schurr, T.G., Cabell, M.F.,
Brown, M.D., Neel, J.V., Larsen, M., Smith, D.G., Vullo, C.M., and
Wallace, D.C. 1993b. Asian affinities and continental radiation of the
four founding Native American mtDNAs. Am. J. Hum. Genet.
53:
563-590.[Medline]
Torroni, A., Miller, J.A., Moore, L.G.,
Zamudio, S., Zhuang, J., Droma, T., and Wallace, D.C. 1994. Mitochondrial
DNA analysis in Tibet: Implications for the origin of the Tibetan
population and its adaptation to high altitude. Am. J. Phys.
Anthropol. 93: 189-199.[CrossRef][Medline]
Torroni, A., Huoponen, K., Francalacci,
P., Petrozzi, M., Morelli, L., Scozzari, R., Obinu, D., Savontaus, M.-L.,
and Wallace, D.C. 1996. Classification of European mtDNAs from an analysis
of three European populations. Genetics
144: 1835-1850.[Abstract]
Torroni, A., Rengo, C., Guida, V.,
Cruciani, F., Sellitto, D., Coppa, A., Calderon, F.L., Simionati, B.,
Valle, G., Richards, M., et al. 2001. Do the four clades of the mtDNA
haplogroup L2 evolve at different rates? Am. J. Hum. Genet.
69: 1348-1356.[CrossRef][Medline]
Tsai, L.C., Lin, C.Y., Lee, J.C., Chang,
J.G., Linacre, A., and Goodwin, W. 2001. Sequence polymorphism of
mitochondrial D-loop DNA in the Taiwanese Han population. Forensic Sci.
Int. 119: 239-247.[CrossRef][Medline]
Voevoda, M.I., Avksentyuk, A.V., Ivanova,
A.V., Astakhova, T.I., Babenko, V.N., Kurilovich, S.A., Duffy, L.K.,
Segal, B., and Shields, G.F. 1994. Molecular genetic studies in the
population of native inhabitants of Chukchee Peninsula. Analysis of
polymorphism of mitochondrial DNA and of genes controlling alcohol
metabolizing enzymes. Sibirskii Ekolog. Z.
1: 139-151.
Watson, E., Forster, P., Richards, M.,
and Bandelt, H.J. 1997. Mitochondrial footprints of human expansions in
Africa. Am. J. Hum. Genet. 61: 691-704.[Medline]
Yao, Y.G., Lu, X.M., Luo, H.R., Li, W.H.,
and Zhang, Y.P. 2000a. Gene admixture in the silk road region of China:
Evidence from mtDNA and melanocortin 1 receptor polymorphism. Genes
Genet. Syst. 75: 173-178.[CrossRef][Medline]
Yao, Y.G., Watkins, W.S., and Zhang,
Y.P. 2000b. Evolutionary history of the mtDNA 9-bp deletion in Chinese
populations and its relevance to the peopling of east and southeast Asia.
Hum. Genet. 107: 504-512.[CrossRef][Medline]
Yao, Y.G., Kong, Q.P., Bandelt, H.J.,
Kivisild, T., and Zhang, Y.P. 2002a. Phylogeographic differentiation of
mitochondrial DNA in Han Chinese. Am. J. Hum. Genet.
70:
635-651.[CrossRef][Medline]
Yao, Y.-G., Nie, L., Harpending, H., Fu,
Y.-X., Yuan, Z.-G., and Zhang, Y.-P. 2002b. Genetic relationship of
Chinese ethnic populations revealed by mtDNA sequence diversity. Am. J.
Phys. Anthropol. 118: 63-76.[CrossRef][Medline]
|
WEB SITE REFERENCES |
http://www.fluxus-engineering.com/;
Network 3.1 program, Fluxus Engineering.
http://www.giib.or.jp/mtsnp/index_e.html;
authors' data.
Received December 17, 2003; Revision received June 14, 2004.
This article has been cited by other
articles: (Search Google Scholar for Other Citing Articles)
|
|

|
|
 ![Home page]()
R. W.
Carter
Mitochondrial diversity within modern human
populations
Nucleic Acids Res., May 14, 2007;
35(9): 3039 - 3045.
[Abstract]
[Full
Text] [PDF]
|
|
|
|
|
.gif)
|
|
 
M.
Tanaka, N. Fuku, Y. Nishigaki, H. Matsuo, T. Segawa, S. Watanabe, K.
Kato, K. Yoko, M. Ito, Y. Nozawa, and Y. Yamada
Women
With Mitochondrial Haplogroup N9a Are Protected Against Metabolic
Syndrome
Diabetes, February 1, 2007; 56(2):
518 - 521.
[Abstract]
[Full
Text] [PDF]
|
|
|
|
|
.gif)
|
|
![J. Med. Genet.]()
R
Hinttala, R Smeets, J S Moilanen, C Ugalde, J Uusimaa, J A M
Smeitink, and K Majamaa
Analysis of mitochondrial DNA
sequences in patients with isolated or combined oxidative
phosphorylation system deficiency
J. Med. Genet.,
November 1, 2006; 43(11): 881 - 886.
[Abstract]
[Full
Text] [PDF]
|
|
|
|
|
|
|
C. H.
Cannon, C. S. Kua, E. K. Lobenhofer, and P.
Hurban
Capturing genomic signatures of DNA sequence
variation using a standard anonymous microarray
platform
Nucleic Acids Res., October 6, 2006;
34(18): e121 - e121.
[Abstract]
[Full
Text] [PDF]
|
|
|
|
|
.gif)
|
|
![Mol Biol Evol]()
M. J.
Pierson, R. Martinez-Arias, B. R. Holland, N. J. Gemmell, M. E.
Hurles, and D. Penny
Deciphering Past Human Population
Movements in Oceania: Provably Optimal Trees of 127 mtDNA
Genomes
Mol. Biol. Evol., October 1, 2006;
23(10): 1966 - 1975.
[Abstract]
[Full
Text] [PDF]
|
|
|
|
|
.gif)
|
|
![Hum Mol Genet]()
Q.-P.
Kong, H.-J. Bandelt, C. Sun, Y.-G. Yao, A. Salas, A. Achilli, C.-Y.
Wang, L. Zhong, C.-L. Zhu, S.-F. Wu, A. Torroni, and Y.-P.
Zhang
Updating the East Asian mtDNA phylogeny: a
prerequisite for the identification of pathogenic
mutations
Hum. Mol. Genet., July 1, 2006;
15(13): 2076 - 2086.
[Abstract]
[Full
Text] [PDF]
|
|
|
|
|
.gif)
|
|
![J. Gerontol. A Biol. Sci. Med. Sci.]()
B. J.
Willcox, D. C. Willcox, Q. He, J. D. Curb, and M.
Suzuki
Siblings of okinawan centenarians share lifelong
mortality advantages.
J. Gerontol. A Biol. Sci. Med.
Sci., April 1, 2006; 61(4): 345 - 354.
[Abstract]
[Full
Text] [PDF]
|
|
|
|
|
|
|

C. Sun,
Q.-P. Kong, M. g. Palanichamy, S. Agrawal, H.-J. Bandelt, Y.-G. Yao,
F. Khan, C.-L. Zhu, T. K. Chaudhuri, and Y.-P. Zhang
The
Dazzling Array of Basal Branches in the mtDNA Macrohaplogroup M from
India as Inferred from Complete Genomes
Mol. Biol.
Evol., March 1, 2006; 23(3): 683 - 690.
[Abstract]
[Full
Text] [PDF]
|
|
|
|
|
.gif)
|
|
![Genetics]()
T.
Kivisild, P. Shen, D. P. Wall, B. Do, R. Sung, K. Davis, G.
Passarino, P. A. Underhill, C. Scharfe, A. Torroni, R. Scozzari, D.
Modiano, A. Coppa, P. de Knijff, M. Feldman, L. L. Cavalli-Sforza,
and P. J. Oefner
The Role of Selection in the Evolution
of Human Mitochondrial Genomes
Genetics,
January 1, 2006; 172(1): 373 - 387.
[Abstract]
[Full
Text] [PDF]
|
|
|
|
|
.gif)
|
|

K K
Abu-Amero, T M Bosley, S Bohlega, and D McLean
Complex I
respiratory defect in LHON plus dystonia with no mitochondrial DNA
mutation
Br. J. Ophthalmol., October 1, 2005;
89(10): 1380 - 1381.
[Full
Text] [PDF]
|
|
|
|
|
.gif)
|
|
![Science]()
V.
Macaulay, C. Hill, A. Achilli, C. Rengo, D. Clarke, W. Meehan, J.
Blackburn, O. Semino, R. Scozzari, F. Cruciani, A. Taha, N. K.
Shaari, J. M. Raja, P. Ismail, Z. Zainuddin, W. Goodwin, D. Bulbeck,
H.-J. Bandelt, S. Oppenheimer, A. Torroni, and M.
Richards
Single, Rapid Coastal Settlement of Asia
Revealed by Analysis of Complete Mitochondrial
Genomes
Science, May 13, 2005; 308(5724): 1034
- 1036.
[Abstract]
[Full
Text] [PDF]
|
|
|
|
|